
凝胶电泳是分离鉴定和纯化DNA片段、检测扩增效果的常用技术方法,通过电泳分离提纯生物大分子的基本原理:由于不同生物大分子的带电性质不同,分子量大小也不一样,在同一电场条件下,其移动方向和迁移率是不同的,因而通过电泳就可把不同类型的生物大分子分开。
操作流程为:1、称样;

2、溶解;
3、加热;
4、倒板;
5、制胶;
6、取样;
7、点样;
8、电泳;
9、检测。
1、 将清洁干燥并经灭菌的eppendorf管(最好0.5ml)编号,用微量移液枪分别加入DNA 1μg和相应的限制性内切酶反应10×缓冲液2μl,再加入重蒸水使总体积为19μl,将管内溶液混匀后加入1μl酶液,用手指轻弹管壁使溶液混匀,也可用微量离心机甩一下,使溶液集中在管底。此步操作是整个实验成败的关键,要防止错加,漏加。使用限制性内切酶时应尽量减少其离开冰箱的时间,以免活性降低。
2、 混匀反应体系后,将eppendorf管置于适当的支持物上(如插在泡沫塑料板上),37℃水浴保温2-3小时,使酶切反应完全。
3、 每管加入2μl 0.1mol/L EDTA(pH8.0),混匀,以停止反应,置于冰箱中保存备用。 1、 取5×TBE缓冲液20ml加水至200ml,配制成0.5×TBE稀释缓冲液,待用。
2、 胶液的制备:称取0.4g琼脂糖,置于200ml锥形瓶中,加入50ml 0.5×TBE稀释缓冲液,放入微波炉里(或电炉上)加热至琼脂糖全部熔化,取出摇匀,此为0.8%琼脂糖凝胶液。加热过程中要不时摇动,使附于瓶壁上的琼脂糖颗粒进入溶液。加热时应盖上封口膜,以减少水份蒸发。
3、 胶板的制备:将有机玻璃胶槽两端分别用橡皮膏(宽约1cm)紧密封住。将封好的胶槽置于水平支持物上,插上样品梳子,注意观察梳子齿下缘应与胶槽底面保持1mm左右的间隙。
向冷却至50-60℃的琼脂糖胶液中加入溴化乙锭(EB)溶液使其终浓度为0.5μg /ml(也可不把EB加入凝胶中,而是电泳后再用0.5μg/ml的EB溶液浸泡染色)。用移液器吸取少量融化的琼脂糖凝胶封橡皮膏内侧,待琼脂糖溶液凝固后将剩余的琼脂糖小心地倒入胶槽内,使胶液形成均匀的胶层。倒胶时的温度不可太低,否则凝固不均匀,速度也不可太快,否则容易出现气泡。待胶完全凝固后拨出梳子,注意不要损伤梳底部的凝胶,然后向槽内加入0.5×TBE稀释缓冲液至液面恰好没过胶板上表面。因边缘效应样品槽附近会有一些隆起,阻碍缓冲液进入样品槽中,所以要注意保证样品槽中应注满缓冲液。
4、 加样:取10μl酶解液与2μl 6×载样液混匀,用微量移液枪小心加入样品槽中。若DNA含量偏低,则可依上述比例增加上样量,但总体积不可超过样品槽容量。每加完一个样品要更换tip头,以防止互相污染,注意上样时要小心操作,避免损坏凝胶或将样品槽底部凝胶刺穿。
5、 电泳:加完样后,合上电泳槽盖,立即接通电源。控制电压保持在60-80V,电流在40mA以上。当溴酚蓝条带移动到距凝胶前沿约2cm时,停止电泳。
6、 染色:未加EB的胶板在电泳完毕后移入0.5μg/ml的EB溶液中,室温下染色20-25分钟。
7、 观察和拍照:在波长为254nm的长波长紫外灯下观察染色后的或已加有EB的电泳胶板。DNA存在处显示出肉眼可辨的桔红色荧光条带。紫光灯下观察时应戴上防护眼镜或有机玻璃面罩,以免损伤眼睛。 经照相机镜头加上近摄镜片和红色滤光片后将相机固定于照相架上,采用全色胶片,光圈5.6,曝光时间10-120秒(根据荧光条带的深浅选择)。
8、 DNA分子量标准曲线的制作:在放大的电泳照片上,以样品槽为起点,用卡尺测量λDNA的EcoRⅠ和HindⅢ酶切片段的迁移距离,以厘米为单位。以核苷酸数的常用对数为纵坐标,以迁移距离为横坐标,在坐标纸上绘出连结各点的平滑曲线,即为该电泳条件下DNA分子量的标准曲线。
9、 DNA酶切片段大小的测定:在放大的电泳照片上,用卡尺量出DNA样品各片段的迁移距离,根据此数值,在DNA分子量标准曲线上查出相应的对数值,进一步计算出各片段的分子量大小(若用单对数坐标纸来绘制标准曲线,则可根据迁移距离直接查出DNA片段的大小)。反之,若已知DNA片段的大小亦可由标准曲线上查出它预计的迁移距离。
10、 DNA酶切片段排列顺序的确定:根据单酶切,双酶切和多酶切的电泳分析结果,对照DNA酶切片段大小的数据进行逻辑推理,然后确定各酶切片段的排列顺序和各酶切位点的相对位置。以环状图或直线图表示,即成该DNA分子的限制性内切酶图谱。
[注意]
1.酶切时所加的DNA溶液体积不能太大,否则DNA溶液中其他成分会干扰酶反应。
2、酶活力通常用酶单位(U)表示,酶单位的定义是:在最适反应条件下,1小时完全降解1mg lDNA的酶量为一个单位,但是许多实验制备的DNA不象lDNA那样易于降解,需适当增加酶的使用量。反应液中加入过量的酶是不合适的,除考虑成本外,酶液中的微量杂质可能干扰随后的反应。
3、市场销售的酶一般浓度很大,为节约起见,使用时可事先用酶反应缓冲液(1×)进行稀释。另外,酶通常保存在50%的甘油中,实验中,应将反应液中甘油浓度控制在1/10之下,否则,酶活性将受影响。
4、 观察DNA离不开紫外透射仪,可是紫外光对DNA分子有切割作用。从胶上回收DNA时,应尽量缩短光照时间并采用长波长紫外灯(300-360nm),以减少紫外光切割DNA。
5、 EB是强诱变剂并有中等毒性,配制和使用时都应戴手套,并且不要把EB洒到桌面或地面上。凡是沾污了EB的容器或物品必须经专门处理后才能清洗或丢弃。
6、 当EB太多,胶染色过深,DNA带看不清时,可将胶放入蒸馏水冲泡,30分钟后再观察。
实验试剂和器材
1.材料:低分子量标准蛋白试剂盒: 低分子量标准蛋白: 兔磷酸化酶B
MW=97,400
牛血清白蛋白
MW=66,200
兔肌动蛋白
MW=43,000
牛碳酸酐酶
MW=31,000
胰蛋白酶抑制剂
MW=20,100
鸡蛋清溶菌酶
MW=14,400
开封后溶于200µl蒸馏水,置-20℃保存,使用前室温融化,沸水浴中加热3-5分钟后上样。 样品1:称3mg样品1,加2 ml蒸馏水溶解。
2.实验试剂
(1)30%丙烯酰胺(Acr):称Acr30g,甲叉双丙烯酰胺
(Bis)0.8g,加蒸馏水至100ml,过滤后置棕色瓶中,
4℃贮存可用1-2月。
(2)10%SDS(十二烷基磺酸钠)
(3)1.5mol/L pH8.8 Tris-HCl缓冲液:称取Tris18.2g,
加入50ml水,用1mol/L盐酸调pH8.8, 最后用蒸馏水定容至100ml。
(4)1.0mol/LpH6.8Tris-HCl缓冲液:称取Tris12.1g,加
入50ml水,用1mol/L盐酸调pH6.8, 最后用蒸馏水定容至100ml。
(5)0.05mol/LpH8.0Tris-HCl缓冲液:称取Tris0.6g,加入50ml水,用1mol/L盐酸调pH8.0,最后用蒸馏水定容至100ml。
(7)10%过硫酸铵(AP)
(8)TEMED(四甲基乙二胺)
(9)样品溶解液:SDS(100mg)+巯基乙醇(0.1ml)+
溴酚蓝(2mg)+甘油(2g)+0.05mol/L pH8.0Tris-HCl(2ml),最后定容至10ml。
(10)固定液:取50%甲醇454ml,冰乙酸46ml混匀。
(11)染色液:称取考马斯亮蓝R250 0.125g,加上述固定液250ml,过滤后备用。
(12)脱色液:冰乙酸75ml,甲醇50ml,加蒸馏水定容至1000ml。
(13)电极缓冲液(内含0.1%SDS,0.05mol/LTris- 0.384mol/L甘氨酸缓冲液pH8.3):称Tris6.0g,甘 氨酸28.8g,加入SDS1g,加蒸馏水使其溶解后定容至1000ml。
实验过程
1.将玻璃板用蒸馏水洗净晾干, 准备2个干净的锥形瓶.
2.把玻璃板在灌胶支架上固定好.※固定玻璃板时,两边用力一定要均匀,防止夹坏玻璃板.
3.按比例配好分离胶,用移液管快速加入,大约5厘米左右,之后加少许蒸馏水,静置40分钟.※凝胶配制过程要迅速, 催化剂TEMED要在注胶前再加入,否则凝结无法注胶.注胶过程最好一次性完成,避免产生气泡.
※水封的目的是为了使分离胶上延平直,并排除气泡 ※凝胶聚合好的标志是胶与水层之间形成清晰的界面.
4.倒出水并用滤纸把剩余的水分吸干,按比例配好浓缩胶,连续平稳加入浓缩胶至离边缘5mm处,迅速插入样梳,静置40分钟. ※样梳需一次平稳插入,梳口处不得有气泡,梳底需水平.
5.拔出样梳后,在上槽内加入缓冲液,没过锯齿时可拆去底端的琼脂糖. ※要使锯齿孔内的气泡全部排出,否则会影响加样效果.
6、加样三个。 (1)取10µl标准蛋白溶解液于EP管内,再加入10µl 2倍样品缓冲液,上样量为20µl。
(2)取10µl样品1溶液,再加入10µl 2倍样品缓冲液,上样量分别为5µl
和10µl。
7.用微量注射器距槽底三分之一处进样,加样前,样品在沸水中加热3分钟,去掉亚稳态聚合。※注射器不可过低,以防刺破胶体,也不可过高,在样下沉时会发生扩散.※为避免边缘效应,最好选用中部的孔注样.
8.电泳槽中加入缓冲液,接通电源,进行电泳,开始电流恒定在10mA,当进入分离胶后改为20mA,溴酚蓝距凝胶边缘约5mm时,停止电泳。
9.凝胶板剥离与染色:电泳结束后,撬开玻璃板,将凝胶板做好标记后放在大培养皿内,加入染色液,染色1小时左右。10.脱色:染色后的凝胶板用蒸馏水漂洗数次,再用脱色液脱色,直到蛋白质区带清晰。※剥胶时要小心,保持胶完好无损,染色要充分.
以上是我实验课件,希望有所帮助!
声明: 我们致力于保护作者版权,注重分享,被刊用文章因无法核实真实出处,未能及时与作者取得联系,或有版权异议的,请联系管理员,我们会立即处理,本站部分文字与图片资源来自于网络,转载是出于传递更多信息之目的,若有来源标注错误或侵犯了您的合法权益,请立即通知我们(管理员邮箱:daokedao3713@qq.com),情况属实,我们会第一时间予以删除,并同时向您表示歉意,谢谢!

 999+
999+ 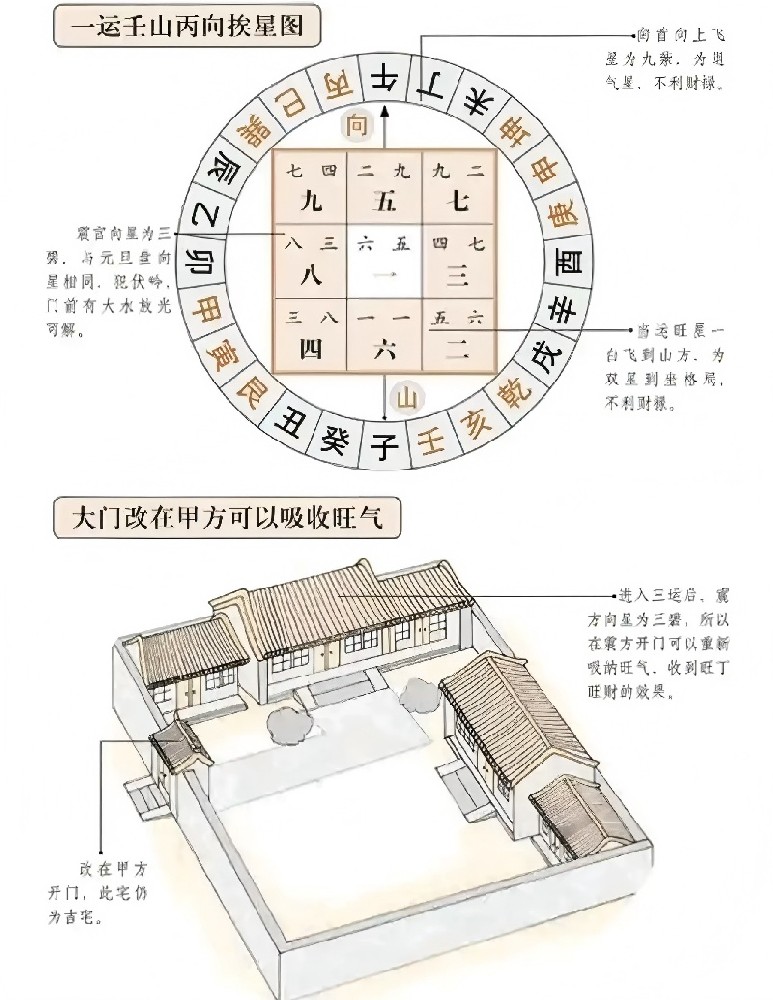 13
13  1843
1843  1922 1576 1961 1481
1922 1576 1961 1481  833 689
833 689  1540
1540 


本站内容仅供参考,不作为诊断及医疗依据,如有医疗需求,请务必前往正规医院就诊
祝由网所有文章及资料均为作者提供或网友推荐收集整理而来,仅供爱好者学习和研究使用,版权归原作者所有。
如本站内容有侵犯您的合法权益,请和我们取得联系,我们将立即改正或删除。
Copyright © 2022-2023 祝由师网 版权所有
邮箱:daokedao3713@qq.com
